
2026年3月10日,《自然材料》(Nature Materials) 在线发表了CAID的短评论文(News & Views)“相变存储材料:解析复杂性”[1],讨论了非晶材料复杂势能面实验测量方面的重要进展[2]。
描述晶体材料势能面较为直观,因为原子是按周期性晶格阵列排列的。而非晶材料(也称为玻璃态材料)缺乏长程周期性有序,其复杂网络结构中的原子之间会产生高度复杂的化学相互作用。因此,非晶材料的势能面通常具有若干能量极大值和极小值。在温度的作用下,非晶状态会在不同的局部极小值之间频繁跃迁。这种非晶弛豫效应会给实际应用带来严重问题。例如,非晶弛豫引发的电阻漂移行为会导致相变存算器件识别精度严重下降。传统势能面的绘制存在较大随机性,而基于实验测量数据绘制非晶势能面则面临极大挑战。
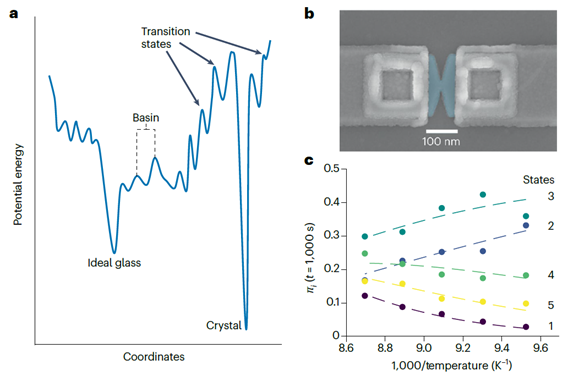
近期,德国明斯特大学Walfort与Salinga等人[2]开发了一种兼具脉冲调制与衬底加热功能的相变存储器件,并基于器件电阻噪声含时/变温的高精度测量与hidden Markov模型拟合,对非晶GeTe材料的结构弛豫过程进行了深入解析。每个电阻状态的平均时长的倒数被定义为状态转变速率,该速率会随着温度升高呈指数增长,并且具有高度的不对称性。以transition state理论与Eyring方程对状态转变速率数据进行拟合,可量化熵与能量对自由能势阱宽度与深度的贡献,进而可以绘制基于实验电学测量的三维势能面地图。该方法仅依赖实验电学测量与数据拟合,并不需要对于特定体系微观转变的前置机理理解,因此预期该方法可应用于其他相关忆阻器材料研究。
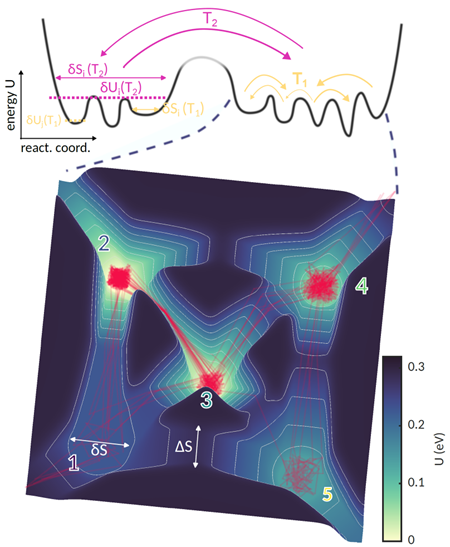
分子动力学模拟等理论方法也可支撑复杂势能面的构建,为非晶弛豫过程提供原子尺度信息。但目前第一性原理分子动力学模拟AIMD的计算效率仅可支撑几纳秒的分子动力学计算。近期,基于机器学习方法研究人员开了适用于相变存储材料精准高效的机器学习势函数。基于该势函数的分子动力学模拟MLMD相较于AIMD而言,计算效率提升了接近6个数量级[3],可支撑微秒量级的分子动力学计算。虽然MLMD计算在时间尺度上仍无法与数十秒至数千秒的实验测量相比较,但其高效的计算过程可结合metadynamics等增强采样方法,加速对势能面进行有效抽样与绘制,从而加强对复杂非晶体系结构弛豫的深入理解。
[1] J.-J. Wang, W. Zhang, Phase-change materials: Unravel the complexity, Nat. Mater. (2026) DOI: 10.1038/s41563-026-02536-3.
[2] S. Walfort, X.T. Vu, J. Ballmaier, N. Holle, N. Vollmar, M. Salinga, A free energy landscape analysis of resistance fluctuations in a memristive device, Nat. Mater. (2026) DOI: 10.1038/s41563-026-02487-9.
[3] Y. Zhou, D.F. Thomas du Toit, S.R. Elliott, W. Zhang, V.L. Deringer, Full-cycle device-scale simulations of memory materials with a tailored atomic-cluster-expansion potential, Nat. Commun. 16, 8688 (2025).
原文链接:https://www.nature.com/articles/s41563-026-02536-3



